INTRODUCTION
In the normal aging process, sleep changes accompany with increasing sleep fragmentation, nighttime awakenings and greater tendency for daytime sleep. Dementia provokes further deterioration of sleep patterns.1 The prevalence of Alzheimer’s disease (AD), most common form, is rapidly increasing and will probably accelerate dramatically within the next decades as a numbers of people are getting older.
Although AD is usually recognized as progressive debilitation of memory, language, and intellect, sleep disturbance is a prevailing and often highly disruptive behavioral symptom associated with AD.2,3 At an early phase of the AD, these neuro behavioral symptoms may appear but seem to be usually associated with a more severe cognitive decline.3,4
The origin of sleep disturbances in AD is considered to be multifactorial. Sleep alterations occur with degeneration of neural pathways that modulate sleep-wake patterns and sleep architecture as well as other environmental factors.3,5 In return, cognitive symptoms may deteriorate through impairment of amyloid-β (Aβ) diurnal pattern and sleep-dependent memory consolidation processes by sleep disorders.6,7 Additionally, a correlation between sleep characteristics and cognitive decline in the elderly has been shown, focusing the fact that sleep and cognition are closely related.8–10
This article will review sleep changes with normal aging and AD, bidirectional relationship between sleep and AD pathology, and underlying mechanism. The result of sleep disturbances in AD will be discussed thereafter. Finally, recommendations as to areas in which future research is needed will be proposed.
SLEEP AND NORMAL AGING
Sleep
Sleep restores the brain and is involved with memory retention. More specifically, slow-wave sleep (SWS) contributes to the consolidation of long-term memory.11 A restorative sleep involves following a diurnal pattern of alertness and activity during the day followed by latency at night. Researchers consider restorative sleep to need cycling several times through the different sleep stages. In the awake state, the electroencephalography (EEG) shows low amplitude, high-frequency fluctuations because neurons in the cerebral cortex fire irregularly. When wakefulness makes way for sleep, the low amplitude, reduction of high frequency activity appear as cortical neurons show a slow oscillation (< 1 Hz) in membrane potential between a hyperpolarized state with no neuronal firing to a depolarized state of intense firing.12 This slow oscillation represents cellular process that organizes wave forms of EEG during sleep, that is, sleep spindles and slow waves.
The EEG background in rapid eye movement (REM) sleep is comparable to the awake state with low amplitude, mixed-frequency activity. Non-rapid eye movement (NREM) sleep consists of 3 stages that are characterized with progressively increased slow wave activity: N1 or drowsiness, N2, and N3 or SWS. During the deeper stages of NREM sleep (N2 and N3), there are more high-amplitude slow waves, which reflect the general decrease in regional synaptic activity during NREM sleep.13 Further, Tononi and Cirelli.14 have found that these periods of deeper NREM sleep reduce synaptic strength to a degree that is energetically sustainable and progression of synaptic plasticity and memory.14
Age-Related Changes in Sleep
Sleep architecture and sleep patterns normally change with aging. The elderly frequently complain of poor sleep including increased sleep latency, difficulty with sleep maintenance, frequent nighttime and early morning awakenings.15 A reduction of total sleep time and sleep efficiency is explained by objective modifications of sleep architecture on polysomnographic studies. Many shifts of sleep stage mark up more frequent nighttime awakenings and sleep fragmentation. These changes can cause excessive daytime sleepiness and increase in diurnal sleep. The number of sleep stages also show alterations: a reduction in SWS and a compensatory increase in the lighter stages of sleep (stage 1 and 2) are shown. Changes covering REM sleep are in significant and seem to advance later with age.15,16 Sleep latency may show similar course with age and microstructural alterations are also found, such as decrease in K complexes and sleep spindles.1 In addition to these structural alterations of sleep, sleep-wake rhythm disturbances have also been depicted in the elderly. An increase in daytime sleep and a tendency to phase advancing are frequent.15 Neuro-hormonal modifications, i.e., melatonin, as well as behavioral factors make circadian sleep and wake rhythm disturbed.17 In elderly, the impact of somatic and psychiatric pathologies, pharmacological treatments, decrease in physical activity and exposure to light should be considered as contributing factors to sleep disturbance. The increasing incidence of specific sleep disorders such as sleep-related breathing disorders, restless legs syndrome and periodic limb movements in sleep has also been observed with aging.15
Sleep Alteration and Cognition in Healthy Aging
In the normal aging elderly, cognitive decline is associated with subjective sleep complaints and objective sleep alteration. Cross sectional and longitudinal studies have reported a correlation between poor sleep quality and cognitive decline.18,19 In mild cognitive impairment (MCI)/dementia in elderly healthy subjects, objective actigraphic-assessed sleep fragmentation and circadian rhythm disruption have been shown as risk factors.9,10 Recently, Westerberg et al.20 delineated sleep disturbances such as reduction of stage 2 sleep spindles and decrease in delta and theta power in amnestic mild cognitive impairment (aMCI) patients. They have observed that an association between a lower subjective sleep quality and a lower recognition of items learned the previous day in a MCI patients.21 It has been still equivocal whether sleep disorders may precede a neurodegenerative process, or may induce cognitive impairment through an increase of depressive symptoms or through alterations in sleep dependent memory consolidation.8,22 Indeed, there are many evidences that sleep optimizes consolidation of different forms of memories.23 While hippocampus-dependent memories (declarative/episodic memories) have been shown to benefit primarily from SWS, memories not depending on the hippocampus (procedural learning related to frontostriatal circuits) build up during periods containing high amounts of REM sleep.24 Almost, these studies about sleep and memory have been performed in young healthy subjects. However, considering that both sleep alteration and cognitive decline are jeopardizing older people, the question of the relationship between sleep and memory consolidation in non-demented and demented elderly is noticeable. Recent studies have suggested an age-related change in the cognitive function of sleep.25,26 The aging effecton sleep-dependent consolidation has mainly been reported that procedural motor sequence learning appears less dependent on sleep periodin older than in younger subjects.27 In case of declarative memories, consequences are more debating: different age groups showed similar sleep-dependent performance changes in on episodic/declarative learning.28,29 However, it has also been reported that episodic memory is less correlated with SWS in health yelderly than in younger adults.30 Thus, sleep-related memory formation partly varies with aging but specific learning functions still get better by sleep, which means a physiological link between aging, sleep (and sleep disorders), and cognition.
Sleep and Preclinical Stage of Alzheimer Disease
Amyloid-β and Alzheimer disease pathogenesis
Alzheimer’s disease is a neurodegenerative disorder which prompts cognitive impairment and causes a current and growing public health crisis that only has limited effective treatments. Age is the greatest risk factor for AD, but genetic (e.g., ApoE4) and environmental (e.g., exercise, diet, and sleep) factors are involved in AD pathology that causes cognitive impairment.31
Studies evaluate the pathological changes, with the earliest identifiable preclinical stage of AD being the accumulation of amyloid plaques in the brain.32
Neurons predominantly produce the Aβ peptide in the brain when amyloid precursor protein (APP) is cleaved by β- and γ-secretase into multiple isoforms of different amino acid lengths.33 Synaptic activity influences the secretion of Aβ isoforms into the interstitial fluid (ISF). Synaptic activity provokes synaptic vesicle exocytosis, which is a significant process involving the secretion of Aβ isoforms.34 The contribution of Aβ isoforms to plaque formation varies. Although Aβ40 is generated at higher concentrations, Aβ42 is more hydrophobic, neurotoxic, and prone to aggregate.35 Moreover, the aggregation of Aβ into extracellular plaques has been observed to be concentration dependent. Studies have also found that aggregation of Aβ in the brain pertains to in soluble amyloid plaques. Extracellular Aβ aggregates into insoluble plaques in the brain. This process has been shown to be a significant early step in the pathogenesis of AD that is associated with the deposition of tau into intracellular neurofibrillary tangles, synaptic dysfunction, neuronal loss, and cognitive impairment.36
These neuropathologic changes in AD are likely to advance in a sequential anatomic pattern involving the entorhinal cortex, hippocampus, and medial temporal lobe.37 In that significant neuronal and synaptic loss precede manifestation of clinical symptoms, Aβ deposition needs further process to result in cognitive decline. Approximately 25–30% of individuals in their eight decade who were cognitively normal have amyloid deposition.38 Preclinical AD is the period of amyloid deposition with normal cognitive functioning.39 In the preclinical phase, Aβ-containing plaques and tau aggregation still do not occur. In the way that significant cell loss leading to the onset of clinically detectable cognitive impairment does not come along in the preclinical phase, treatment of AD in this phase is considered proper.40
In the aggregation of Aβ and formation of amyloid plaques, synaptic activity occupies a consequential role. These synaptic activities are impacted by the state of brain network. The default mode network (DMN) is the resting state network most active in the absence of task performance41 and AD has been reported to reach the DMN first.42 The DMN is correlated with episodic memory,43 which is a cognitive domain that deteriorates early in AD. Because the DMN is a resting state network and is therefore synaptically and metabolically more active than other regions of the brain, this co-localization with AD pathology may be results of increased Aβ production and secretion into the ISF. Thereafter, amyloid deposition leads to decreased energy metabolism and atrophy in that brain region.44
Sleep and Alzheimer Disease Pathogenesis
Diurnal pattern of amyloid-β in cognitive impairment in elderly
In both mice45 and humans,46 a degree of soluble Aβ fluctuates with the sleep-wake cycle, in a diurnal pattern. A potential mechanism, through which sleep problems may increase the possibility of developing AD, is inferred by this observation.
In a case of animal model, Kang et al.47 found that APP transgenic mice (Tg2576) with amyloid deposition show an association between the sleep-wake cycleand brain ISF Aβ concentrations, and soluble Aβ levels in the ISF of APP transgenic mice appear a diurnal pattern. While these mice have normal sleep when they are young before amyloid deposition, once Aβ plaque formation in APP transgenic mice advances, there is a deprivation of the Aβ diurnal pattern. Further, sleep-wake cycle is disrupted dramatically with increased wakefulness and decreased sleep during the light phase when the mice would be expected to sleep.48
In a report of humans, researchers collected serial cerebrospinal fluid (CSF) samples via lumbar catheter every hour for 36 hours and at the same time monitored sleep with ambulatory polysomnography and observed activity levels with video to explore the possible correlation between fluctuating Aβ concentrations and time of day or activity levels.46 There was a fluctuation by 25% in both soluble Aβ40 and Aβ42 isoform concentrations over the collection period and these fluctuations fit a cosine wave, which is consistent with a diurnal pattern. Although soluble Aβ levels varied with the sleep-wake cycle after a 6-hour delay, no correlation between Aβ fluctuation and activity levels was found.46 Moreover, the amplitude of the cosine wave decreased with age from young normal controls to individuals > 60 years who were Pittsburgh compound B-positron emission tomography negative and the amplitude decreased more in individuals > 60 years who were PiB-PET positive for amyloid deposition. The Aβ diurnal pattern was also observed in individuals with and without autosomal dominant AD mutations (e.g., presenilin-1 and presenilin-2).
Effect of Alzheimer disease pathology on sleep
Amyloid-β aggregation would directly debilitate neuronal function in brain regions critical to sleep and wake promotion.37 In studies of APPswe/PS1δE9 mice, which are manipulated to develop amyloid plaques, sleep-wake cycles and Aβ diurnal variation became aberrant following the onset of amyloid deposition.48 When active immunization with Aβ42 eliminated amyloid plaques, sleep-wake cycles returned to normal. These data strongly suggest an evidence to a direct causal role of amyloid deposition in disrupting sleep-wake mechanisms.
Another studies in mouse models of AD confirm the correlation between AD pathology and disrupted sleep, and support a possible causal relationship. In one transgenic mouse model (the amyloid precursor protein/presenilin 1 or APP/PS1 model), disrupted sleep patterns become evident with the increasing extent of Aβ deposition in the brain. Further, reduced sleep duration, slowed EEG during wakefulness, shorter sleep bouts and reduced circadian amplitude are detected in the APP/PS1 knock-in model of AD and the PLB1 triple knock-in model (in which mice carrying a single copy each of mutant human APP and tau transgenes are crossed with mice over expressing PS1).49
In a case of humans, individuals with preclinical AD who are cognitively normal, but have biomarker evidence of amyloid plaques show worse quality of sleep, as assessed by actigraphy-measured sleep efficiency and wake time after sleep onset than groups who do not have evidence of amyloid plaques.50 These differences are persistent even after adjustment for age, sex, and the presence of the apolipoprotein E (APOE) ɛ4 allele (an important risk factor for late-onset, sporadic AD). The importance of this finding is that although all individuals participating were cognitively normal, sleep parameters were separated in these individuals by amyloid deposition status. Thus, this research supports the hypothesis that sleep disturbances are associated with AD pathology.
Although further research is required to tease apart the contributions of these and other factors to sleep problems in AD, the data from preclinical AD in humans and mouse models reinforces a direct negative effect of Aβ accumulation on sleep function.
Effect of sleep on Alzheimer disease pathology
Some mechanisms have been suggested, by which poor sleep may contribute to amyloid deposition. Soluble Aβ is secreted during physiological synaptic activity.34 Since synaptic activity is correlated with sleep and awake state, there is a difference in Aβ secretion between sleep and wakefulness. This diurnal variation insoluble Aβ has been demonstrated in several studies in miceas well as in humans to contribute to sleep-wake states, though some aspect of circadian factors has not been entirely ruled out.46,47 Thus, brain regions with the highest soluble Aβ levels are also most vulnerable to the formation of amyloid plaques.51 Notably, in a quiet awaken state, brain regions in the DMN41 are the most active areas. Therefore, brain regions in the DMN are most apt to shape up the amyloid deposition during the development of AD pathology.52 Based on these data, a presumptive mechanism would be that chronically insufficient sleep causes relatively increased neuronal activity and, therefore, a relative excess of soluble Aβ. Indeed, prospective studies have shown poor sleep quality as a risk factor for cognitive decline.22
In contrast to DMN, NREM sleep is a relatively quiescent state at the neuronal level. The period of SWS is the deepest stage of NREM sleep in which glucose utilization measured by 18F-fluorodeoxyglucose PET studies decreases by 43.8% compared with the awake state and NREM and REM sleep period.53,54 Specifically, cortical neurons continuously fire irregularly in both the awake and REM states, representing low-amplitude, high-frequency waves on EEG. During SWS, cortical neurons fluctuate between silent periods of hyperpolarization and firing during depolarization. This oscillation appears on EEG as high-amplitude, low-frequency waves, or SWS.55
The quality of sleep reflects the amount of time during the sleep period that cortical neurons will depolarize and fire. Indeed, acute sleep deprivation in mice led to an increase in the concentration of soluble Aβ independent of the stress-response pathway, as depicted by blocking corticotropin-releasing factor.47 Additionally, Spira et al.56 has found that poor sleep quality in older adults was correlated with increased brain levels of Aβ, a well-known AD biomarker. Further, Andrew Lim et al.57 showed that AD incidence and the degree of AD pathology in individuals with the ɛ4 allele of APOE are decreased by unfragmented sleep. Even among APOE ɛ4 carriers, a subset of individuals with excellent sleep consolidation are found to be at a reduced risk of AD, and there are also a subset of individuals with very poor sleep who may be at a particularly high risk of AD.
Connecting with the findings in humans, Nedergaard and colleagues disclosed the neuropathological mechanisms by which sleep disturbances are linked to cognitive impairment through describing the important step. These researchers delineated the function of the brain glymphatic system, which clears interstitial proteins through recirculation of CSF, which interchanges with ISF. They postulated that the sleep-wake cycle influences energy-consuming transport of fluids and soluble molecules. Nedergaard’s team used fluorescent markers and two-photon imaging in awake, sleeping and an aesthetized mice to track the transport of brain ISF and Aβ peptides. Sleeping and an aesthetized mice showed the larger interstitial space in the brain by 60% than in awake mice. Increased exchange between CSF and ISF in sleeping and an aesthetized mice permitted more-efficient clearance of neurotoxins from the brain during sleep.58
Together, these studies explored the mechanisms that link sleep disturbance to AD pathophysiology, and suggested that the important role of sleep might be its ability to ameliorate clearance of metabolic waste products from the brain.
Bidirectional relationship between sleep and Alzheimer disease pathology
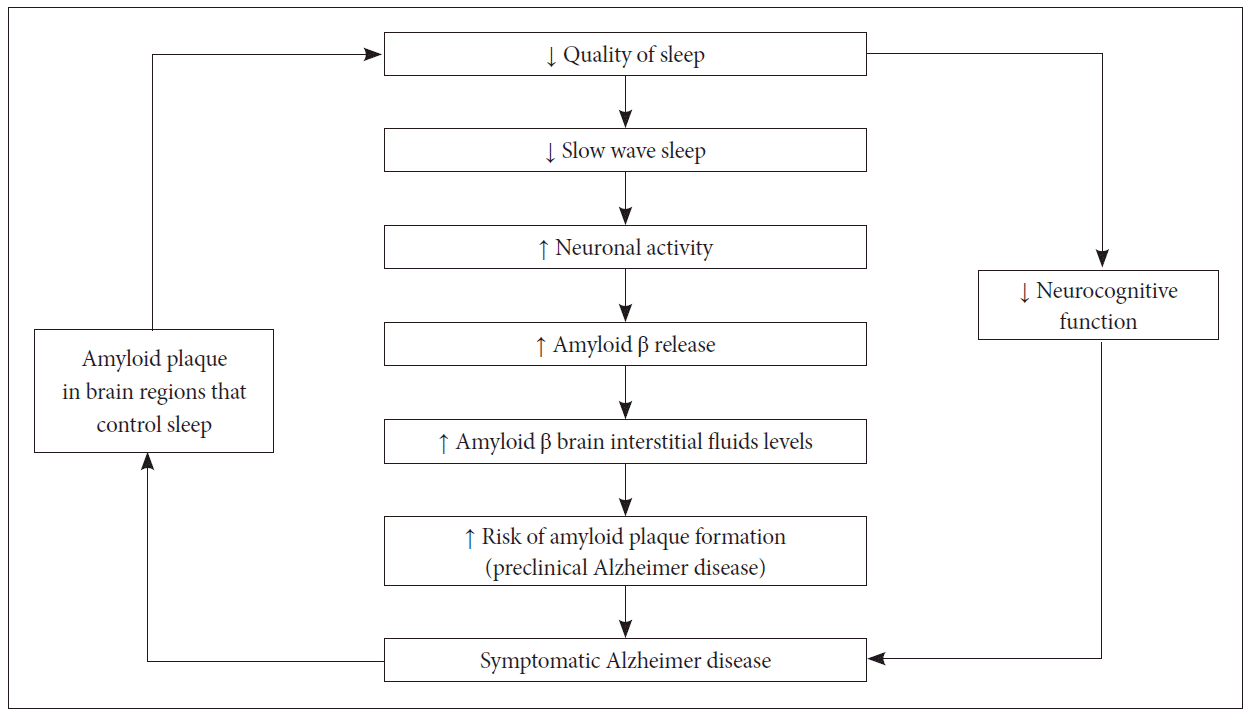
By putting above studies together, we can premise that Aβ accumulation negatively impacts sleep-wake behaviors, and conversely, poor quality of sleep may increase risk of Aβ aggregation. This bidirectional relationship can be explained as positive feedback loop and associated factors that influence this relationship. For instance, obstructive sleep apnea may cause hypoxic stress and inflammation and increase Aβ levels via increased wakefulness. Thus, Aβ advances through these effects. Cognitive and physical activity levels have a bidirectional relationship with both sleep-wake patterns and AD, thus may amplify the feedback loop between poor sleep and AD.
That is, impaired cognitive function is correlated with sleep disturbances, feeding back to the sleep changes associated with aging. This process results in increasing Aβ deposition that is a known marker for AD risk (Fig. 1).
Indirect factor contributing to sleep disturbance in cognitive impairment
Indirect factors can make disrupted sleep-wake in AD and aging persistent. Specifically, preclinical amyloid deposition is associated with decreased physical activity,59,60 and because exercise accentuates sleep, lack of physical activity may lead to a reduction of sleep quality. Further, depression, a frequent and early symptom of dementia,61 often appears in insomnia. Cognitive changes precipitate retirement from work, which may cause irregular activity and sleep patterns. In later phase of dementia, institutional settings are associated with insufficient exposure to light and activity. Therefore, this consequence is associated with further deterioration in circadian rhythms andsleep-wake patterns.45
Elderly with AD patients suffer from medicalas well as psychiatric morbidities, which can disrupt the sleep-wake cycle. These include pain (e.g., from arthritis), nighttime cough, dyspnea (from cardiac or pulmonary illness), gastroesophageal reflux, in continence/frequent nighttime urination and depression. Drug combination therapy may affect sleep quality either by disrupting nighttime sleep as do diuretics and stimulating agents (e.g., sympathomimetics, bronchodilators) or by sedating effect (e.g., antihistamines, anticholinergics, sedating anti depressants) which prompts daytime sleepiness. All these exogenous factors, which require non-pharmacological treatments to reverse their harmful effects, can easily be managed by the medical staff.
SLEEP AND ALZHEIMER DISEASE
Sleep Architecture Alterations
Sleep disturbances in patients with AD are qualitatively comparable to those seen in the normal elderly, but the degree of sleep disturbances is much more severe, excluding REM sleep which alters with dysfunction in cholinergic neurotransmission.
Total sleeptime reduction appears due to increasing wakefulness after sleep onset as AD progresses and, together an extension in sleep latency.1,62 Clinically, this disorder influences agitation during the night and/or somnolence during the day.
Because these patients show to pographically diffuse and low-amplitude slow-wave (0.5–2 Hz) activity representing both the sleeping and waking EEGs, it is difficult to differentiate among NREM sleep stages, and to distinguish waveforms characterizing SWS in particular in the AD patient. Some researchers have characterized NREM sleep in AD patients as in determinate by observing this delta activity, called frontal intermittent rhythmic delta activity.62 Despite some disparities, most researchers affirm a decrease in SWS and alterations in spindles and K complexes.63,64
While REM sleep is relatively intact in normal aging REM sleep, it is quantitatively reduced in AD. However, the total number of REM sleep episodes and REM sleep latency remain invariant.64 One of biological markers of AD is an EEG slowing during REM sleep.63 Additionally, Hassainia et al.65 found an increase in absolute delta and theta activities, and a decrease in absolute alpha and beta activities during REM sleep in AD, which appear particularly in the parietotemporal and frontal regions.65
Lastly, when AD patients perform specific tasks, they have shown modifications of sleep. Studies reported a faster mean theta frequency in both REM sleep and SWS during post-learning sleep in AD patients comparing with elderly controls. Since this change in theta rhythm was correlated with better delayed episodic recall, it is suggested that this electrophysiological feature could reflect compensatory functions to maintain memory performance.66 Similar with this result, a correlation between specific decrement in fast spindles and learning abilities has also been described.67
Sleep-Wake Cycle Alteration
Sundowning syndrome, which modifies a sleep-wake cycle, is a particularly stressful phenomenon for the patient and caregivers and characterized by the emergence or an increment of neuropsychiatric symptoms suchas confusion, agitation, anxiety, and aggressiveness in the late afternoon, evening, or at night. The severity of cognitive impairment is associated withthe progress of sundown syndrome.68 Circadian rhythm deteriorations in AD concern other circadian rhythms such as body core temperature, motor activity and several hormone secretions as well as sleep/wake cycle.
Despite AD pathology does not influence the pineal gland, a reduction in CSF melatonin level, which potentially reflects alteration of the suprachiasmatic nucleus output, has been explored in these patients.69 Variation in the melatonin secretion rhythm has also been observed.70 These findings explain impairment of the endogenous pacemaker, which may cause a reduction of capacity to synchronize physiological rhythms.71 Exogenous factors do a critical role in disturbing a sleep/wake rhythm. External zeitgebers are important in the maintenance of circadian rhythmicity in humans by interacting with the central clock. During AD, the cognitive decline and associated visual neurosensory dysfunctions lead to decreased input to the suprachiasmatic nucleus, thus reduced zeitgebers (such as light, diurnal activities) and may conclusively play an important role in debilitating the sleep-wake cycle.72
Risk Factors for Sleep Disorders in Alzheimer Disease
Severity of the illness
Although sleep disorders may be present in an early stage of the illness, many studies suggest that they are aggravated over time. In aspecial careunit, Bliwise et al.73 depicted that severe dementia was associated with more sleep disturbance. Yesavage et al.74 reported significant deterioration of nocturnal sleep parameters measured by longitudinal actigraphy over the course of approximately 1.5 year follow-up. Additionally, studies have described an association between daytime napping and the severity of cognitive decline in AD patients and objective polysomnographic recordings confirm the relationship between mini-mental state examination and mean daytime sleep latency in mild/moderate AD patients.75,76 However, some studies show discrepancies with those results. It has been recently reported that there is an absence of correlation between sleep score (Neuropsychiatric Inventory) and disease severity.77 Furthermore, sleep disturbance does not impact all patients with AD and the stage of the cognitive impairment seems to reflect only a small part of the alteration regarding the presence of sleep/wake disorders.78 This could support an individual and potentially genetically determined susceptibility.
Genetic susceptibility
The ApoE4 allele is a known risk factor for advancing AD, especially in homozygous cases.78 A relation between the presence of sleep disturbance and APOE (a protein involved in maintenance and remodeling of neuronal membranes) isoform has also been mentioned. Most studies correlate the ApoE4 allele with sleep alterations (e.g., reduction of REM sleep) in MCI or demented patients.79 Alterations in the production of melatonin could cause increased sleep disturbances in ApoE4 patients: Liu et al.80 have evidenced alower CSF melaton in rate in homozygous 4/4 than in heterozygote patients. Moreover, researchers have associated genotype 4/4 with an increment of risk in obstructive sleep apnea and hypopnea (OSAH) patients, and with sleep alterations and cognitive impairment in patients with OSAH.81–84 Despite, some authors have reported debatable results: Yesavage et al.85 have reported a protective effect of ApoE4 allele on sleep in AD patients, and Craig et al.86 have depicted no effect of ApoE genotype. Further studies need to explore a more complex relationship between ApoE4 status and sleep to explain these contradictory observations.
Indeed, a better sleep consolidation could reduce the increased risk conferred by the ApoE genotype on the development of AD.57 Besides ApoE studies, monoamine A oxidase promoter polymorphism, which may affect sleep/wake regulation by affecting the availability of serotonin, a precursor of melatonin, has been investigated; Craig et al.86 showed that the high-activity 4-repeat allele of the monoamine A oxidase variable number tandem repeat promoter polymorphism conferred increased vulnerability to sleep disturbance. Moreover, allelic variations in this gene have been correlated with behavioral and personality traits in other contexts, which may clarify this association.87
COGNITIVE CONSEQUENCES OF SLEEP DISTURBANCES IN AD
A significant one of clinical neuropsychiatric symptoms of AD is a sleep disorder. Despite the critical role of sleep in memory consolidation, alterations of sleep in AD patients may be described in itself as an aggravating factor for amnestic symptoms.6
In AD patients, sleep alters more profoundly than in healthy elderly and acorrelation between sleep architectural modifications and learning performances has been reported. Rauchs et al.67 found that AD patients with the decrease in fast sleep spindles during post-learning sleep showed poorer immediate recall performance in an episodic memory task, and the ability to retrieve recent autobiographical memories was positively associated with the amount of SWS.88 The variation of REM sleep, reflecting a decrease of acetylcholine levels, may also play a role in memory deficits in AD patients. Additionally, experimental pharmacological REM sleep has been augmented with cholinesterase inhibitors and has appeared to do positive effects on procedural memory consolidation.89
Indeed, cognitive function in AD patients deteriorates from the start of short term memory. However, other cognitive functions, such as language, motor skills, and attention are also impaired. In AD, besides learning and memory, executive function and emotional reactivity might be attenuated by sleep disturbances, too. AD patients also experiment circadian rhythm disruption, which could in itself lead to cognitive impairment. Several studies have supported this hypothesis. Experimental procedures in animals allow distinguishing age-related from disrupted rhythms-related cognitive alterations. In hamsters, Antoniadis et al.90 reported that circadian rhythm fragmentation weakens ability to form cognitive associations. In rats, they found a deleterious effect of repeated phase shifts on hippocampal (spatial) memory.91 In humans, circadian rhythm sleep disorders have often been known to impair selective attention and executive function.92
CONCLUSION AND FUTURE DIRECTIONS
Sleep disturbances are common in AD and have an important effect on patients and caregivers. In regard of sleep disruption increasing risk of future AD, it has a critical value to confirm and treat individuals with sleep disorders, such as obstructive sleep apnea and to explore whether good quality of sleep in humans can either mark down the risk of AD or slow down the progression of preclinical to symptomatic AD. Studies of individuals with both amyloid deposition and consequent neuronal injury such as elevated CSF tau levels and amyloid plaques will promote the research on the correlation between sleep and pathological progression in AD. Lastly, further studies will need to confirm evidence-based guidelines regarding both sleep evaluation and treatment in this growing population.